Due to the health emergency of Covid19, on Thursday 23 April 2020 the European Council approved and formalized the one-year extension of the 2017/745 Regulation on medical devices, postponing their application until 26 May 2021 (https://data.consilium.europa.eu/doc/document/ST-7180-2020-INIT/en/pdf). What research areas are involved, and what it foresees this legislation, for a specific type of device, we tell it in this article, proposing our patented solution.
Recently the Regulation that affects medical devices has been revised: the Parliament and the European Council have approved the following two regulations amending the existing ones. We are talking about the EU regulation 2017/745, relating to medical devices (MDR, Medical Device Regulation), and the EU regulation 2017/746, relating to in vitro diagnostic medical devices (IVDR, In Vitro Diagnostic Regulation).
The EU Regulation 2017/745, in fact, entered into force on 25 May 2017, but a transition period of three years has been foreseen, which, due to the health emergency, have become four, for which it will become fully operational from May 26, 2021, and companies in the medical device sector will have to adapt by that date to the new requirements foreseen by the regulation in carrying out their activities. The extension does not concern the 2017/746 Regulation on in vitro diagnostic medical devices, whose application remains scheduled for May 26, 2022.
But what is a medical device?
Article 2 of Regulation 2017/745 defines a medical device as “any instrument, appliance, equipment, software, plant, reagent, material or other article, intended by the manufacturer to be used on humans, alone or in combination, for one or more of the following specific medical uses:
– diagnosis, prevention, monitoring, forecasting, prognosis, treatment or mitigation of diseases,
– diagnosis, monitoring, treatment, alleviation or compensation of an injury or disability,
– study, replacement or modification of the anatomy or of a physiological or pathological process or state,
– provide information through the in vitro examination of samples from the human body, including blood and donated tissues, and which does not exercise in or on the human body the main action for which it is intended by pharmacological, immunological or metabolic means, but whose function can be assisted by these means.
The specific characteristics of a medical device are the therapeutic and / or diagnostic medical purpose and the main mechanism of non-pharmacological, neither immunological, nor metabolic action.
Among the medical devices, there are those based on substances, or all those devices consisting of solutions, suspensions, ointments, sprays, tablets, bars, drops, powders that are applied on the body or introduced into it, whose main action is purely medical, obtained with a physical, mechanical, chemical-physical mechanism, clinically evaluated, but they can also have an ancillary action.
In Regulation (EU) 2017/745 these invasive devices are considered as a unique group, and their classification is expressed by rule 21 of Annex VIII.
Rule 21 states that: “devices consisting of substances, or combinations of substances, intended to be introduced into the human body through an orifice of the body, or to be applied to the skin and which are absorbed by the human body, or locally dispersed in it include:
- In class III if they, or their metabolism products, are absorbed systemically by the human body in order to achieve their intended use;
- In class III if they achieve their intended use in the stomach or lower gastrointestinal tract and they, or their metabolism products, are absorbed systemically by the human body;
- In class IIa if they are applied on the skin or if they are applied in the nasal or oral cavity up to the pharynx and achieve their intended use on said cavities and
- In class IIb in all other cases.
This legislation, in fact, involves all companies related to the chemical sector of medical devices. If previously it was only the pharmaceutical companies that had to subject the new products to testing, with the new provisions, all companies are obliged to operate according to a correct risk assessment and a clinical evaluation of the product before it is placed on the market.
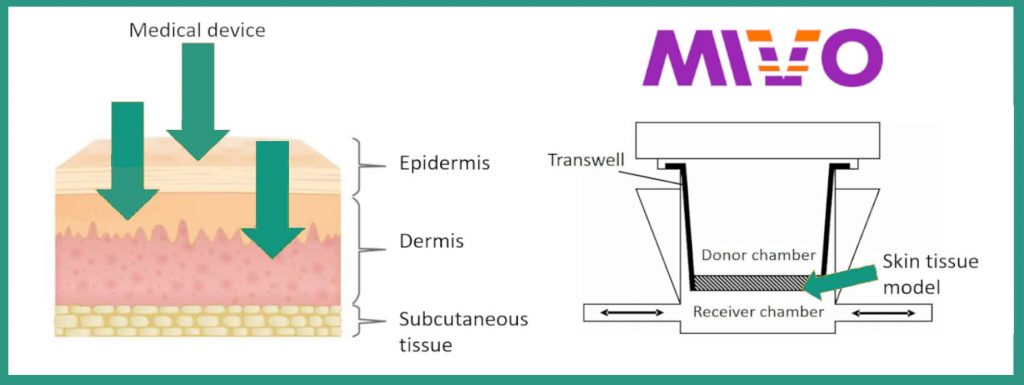
 To obtain the classification envisaged in European legislation, it is therefore necessary to subject the future medical device to an absorption test. The concept of absorption is therefore added to the concept of intended use: it becomes central for classification, and does not provide for intermediate rules.
To obtain the classification envisaged in European legislation, it is therefore necessary to subject the future medical device to an absorption test. The concept of absorption is therefore added to the concept of intended use: it becomes central for classification, and does not provide for intermediate rules.
This legislation, in fact, involves all companies related to the chemical sector of medical devices. If previously it was only the pharmaceutical companies that had to subject the new products to testing, with the new provisions, all companies are obliged to operate according to a correct risk assessment and a clinical evaluation of the product before it is placed on the market.
To obtain the classification envisaged in European legislation, it is therefore necessary to subject the future medical device to an absorption test. The concept of absorption is therefore added to the concept of intended use: it becomes central for classification, and does not provide for intermediate rules.
React4life has implemented a protocol to carry out absorption tests on the medical device by recreating the physiological environment in vitro.
Absorption critically determines the bioavailability of the compound. In order for a compound to reach a tissue, it usually needs to be included in the bloodstream – often through mucous surfaces such as the digestive tract (intestinal absorption) – before it is absorbed by the target cells. Factors such as poor solubility of the compounds, gastric emptying time, intestinal transit time, chemical instability in the stomach, and inability to permeate the intestinal wall allow to reduce the degree of absorption of a medical device after oral administration.
The innovative solution proposed by React4life, for the in vitro absorption test, involves the use of a patented technology, which consists of a multi-chamber fluidic device, the MIVO, which, among its advantages, has that of being compatible with most epithelial tissues (e.g. skin, vaginal wall, oral mucosa) commercially available or recreated in the laboratory through cell cultures. By connecting the MIVO with a peristaltic pump or syringe, it is possible to reproduce in vitro the systemic circulation that feeds the tissue and that influences the absorption kinetics that characterizes the real condition in vivo. Specifically, the MIVO is composed of a donor department, which houses the medical device to be tested, and a receiving compartment, which receives the quantity that has passed through the tissue, after a given interval of time. All in a fluid-dynamic regime, as dynamic are the stimuli in which cells and tissues are constantly subjected.